- CARBOXYLIQUES (ACIDES)
- CARBOXYLIQUES (ACIDES)Parmi les acides organiques, caractérisés par la propriété de former un sel avec la soude ou la potasse, les acides carboxyliques, définis par le groupement carboxyle fonctionnel 漣C2H, tiennent une place prépondérante.Le plus anciennement connu est l’acide acétique CH3C2H (acetum , vinaigre), qui prend naissance par oxydation enzymatique des solutions diluées d’alcool éthylique, du vin par exemple. Il a été longtemps préparé par pyrolyse du bois à l’abri de l’air: il se forme un mélange complexe appelé pyroligneux dont les principaux constituants de la partie liquide sont le méthanol et l’acide acétique.Le premier représentant de la fonction, l’acide formique HC2H, doit son nom à sa présence chez la fourmi rouge, dont on l’a tiré autrefois.De très nombreux acides carboxyliques existent soit à l’état libre, soit le plus souvent à l’état d’esters, dans les produits naturels les plus divers: corps gras des huiles végétales et graisses animales, lipides des structures cellulaires, cytoplasmiques et membranaires. L’extraction des acides gras (à longue chaîne carbonée paire) des corps gras naturels est pratiquée de longue date par hydrolyse à chaud, ou saponification à la soude, suivie d’une acidification par l’acide sulfurique: le glycérol est facilement séparé des acides qui se solidifient au refroidissement. Les savons ou sels alcalins des acides gras étaient connus des Gaulois qui les utilisaient, semblet-il, comme cosmétiques.1. NomenclatureLa nomenclature officielle I.U.P.A.C. (International Union of Pure and Applied Chemistry) des acides carboxyliques est obtenue en remplaçant le e terminal de l’hydrocarbure correspondant par le suffixe -oïque et en faisant précéder le nom du terme acide : acide alcanoïque. Le numérotage est inutile, la fonction acide ne pouvant être portée que par un carbone primaire. Si l’acide est ramifié, on choisit comme chaîne principale la plus longue de celles qui portent la fonction. Lorsque cette nomenclature est compliquée, on désigne la fonction par le mot carboxylique utilisé comme suffixe: acide cyclohexanecarboxylique. Cependant, certains noms vulgaires, rappelant souvent l’origine de l’acide, restent en usage.2. État naturel et préparationsLes corps gras d’origine animale et végétale sont, en très grande majorité, constitués par des esters d’un trialcool, le glycérol, et d’acides linéaires à nombre pair d’atomes de carbone, éventuellement insaturés ou porteurs d’une fonction alcool, appelés acides gras (glycérides). Les cires, animales et végétales (cérides), sont des esters d’acides gras et d’alcools à longue chaîne: le blanc de baleine ou palmitate de cétyle est l’ester de l’acide palmitique C15H31C2H et de l’alcool cétylique C15H31CH2OH (hexadécanol), tandis que la cire de Carnauba renferme en particulier du palmitate de myricyle (C15H31C2C30H61). Les stérides, esters d’acides gras et d’alcools particuliers appelés stérols, sont également présents à l’état naturel: la lanoline, extraite de la graisse de suint de mouton qui est un ester de lanostérol (un alcool tétracyclique en C30), les stérides du plasma sanguin.Qu’elles soient industrielles ou de laboratoire, les principales méthodes de préparation des acides carboxyliques peuvent être classées en deux familles, selon que la chaîne carbonée du substrat de départ est conservée ou modifiée.Réactions de préparation sans modification de la chaîne carbonéeHydrolyseLes réactions d’hydrolyse des esters naturels, notamment des triglycérides provenant des graisses animales, sont réalisées en présence de soude et donnent, outre le glycérol, un mélange de sels de sodium des acides palmitique, stéarique et oléique. Ce savon de sodium peut être hydrolysé en milieu acide (H2S4) et fournit les acides eux-mêmes (réaction 1).L’hydrolyse des fonctions trivalentes, obtenues par synthèse, donne également accès aux acides carboxyliques. Les nitriles en particulier, facilement accessibles par action du cyanure de potassium sur les halogénures ou les tosylates d’alkyle, sont hydrolysés en catalyse acide ou basique. Les dérivés trichlorométhylés des aromatiques, résultant de la chloration photochimique des dérivés méthylés correspondants, sont hydrolysés en catalyse acide en acides carboxyliques.OxydationL’oxydation des alcools primaires en aldéhydes puis en acides est réalisée, au laboratoire, sous l’action d’oxydants énergiques comme KMn4 ou K2Cr27 en milieu acide (cf. ALCOOLS, dérivés CARBONYLÉS). L’une des préparations industrielles de l’acide acétique concerne l’oxydation, par l’air, de l’acétaldéhyde, avec catalyseur au cobalt, à température ordinaire et sous faible pression.Les cétones cycliques sont oxydées, avec ouverture de la chaîne, en diacides: c’est l’un des procédés d’obtention de l’acide adipique par oxydation de la cyclohexanone (cf. dérivés CARBONYLÉS).Enfin, les hydrocarbures aromatiques méthylés, notamment le toluène, peuvent être oxydés par l’air, en présence de catalyseurs au cobalt, en acides arylcarboxyliques.Réactions de préparation avec augmentation de la chaîne carbonéeCarboxylation d’organométalliquesUn procédé très général de synthèse des acides carboxyliques est l’addition des organomagnésiens sur l’anhydride carbonique. D’autres dérivés de l’acide carbonique peuvent être utilisés, notamment le chloroformiate et le carbonate d’éthyle, qui conduisent à l’ester éthylique de l’acide à un carbone de plus.La synthèse malonique est également une méthode très générale pour introduire, sur une chaîne alkyle ou aryle, un groupement 漣CH2C2H. Le malonate d’éthyle est transformé en carbanion par une base forte comme l’éthylate, ou, mieux, l’amidure de sodium. L’halogénure d’alkyle ou d’aryle est condensé sur ce carbanion et conduit à l’alkyl- ou aryl-malonate d’éthyle. Par hydrolyse acide, on passe à l’acide malonique substitué qui, comme tous les 廓-diacides, se décarboxyle par chauffage en donnant, sous sa forme énolique, l’acide à deux carbones de plus que le substrat de départ (réaction 2).Hydroxycarboxylation d’alcènesDeux procédés industriels mettent en œuvre l’addition, sur des oléfines, de CO + H2O:– le premier, dû à Reppe, est surtout intéressant au départ de l’éthylène: par action de CO + H2O à 300 0C sur un catalyseur de nickelcarbonyle, on obtient l’acide propionique ;– le second, dû à Koch, est plus général et permet en particulier la synthèse d’acides encombrés en 見. Il fait agir, en présence d’un catalyseur acide, une oléfine ramifiée avec le mélange CO + H2O; à partir de l’isobutène, on obtient l’acide pivalique:
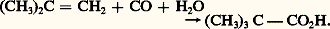 L’hydroxycarbonylation de la soude est l’une des voies industrielles d’accès à l’acide formique. Une variante de ce procédé remplace l’eau par le méthanal et conduit au formiate de méthyle qu’une ammonolyse et une hydrolyse irréversible transforment en formamide et acide formique.L’hydroformylation des oléfines [cf. ALCÈNES], suivie de l’oxydation des aldéhydes formés, est également une voie d’accès aux acides carboxyliques (acides propionique et butyrique). Le principal mode de fabrication de l’acide acétique est aujourd’hui la carbonylation du méthanol (cf. acide ACÉTIQUE).Réactions de préparation avec dégradation de la chaîne carbonéeOxydationDe nombreuses réactions d’oxydation, avec coupure de la chaîne carbonée, conduisent à des acides, stade ultime de l’oxydation ménagée des molécules carbonées.Des procédés industriels, fondés sur l’oxydation, sans catalyseur, d’hydrocarbures, permettent de préparer des mélanges, parfois complexes, de produits d’oxydation où les acides carboxyliques sont majoritaires. Le principal problème est celui de la séparation [cf. ALCANES].Au laboratoire, la coupure oxydante des alcènes, par le permanganate ou le bichromate acides, conduisent à des mélanges d’acides. L’ozonolyse des alcènes est une méthode plus analytique que préparative.Plus intéressante est la coupure oxydante des cétones dans des conditions contrôlées:– coupure des méthylcétones par les halogènes en milieu alcalin. Il se forme une trihalométhylcétone (cf. dérivés CARBONYLÉS) qu’un chauffage en milieu basique transforme en haloforme et sel alcalin de l’acide ayant un carbone de moins que la méthylcétone de départ;– oxydation, avec réarrangement, des dérivés carbonylés sous l’action des peracides (réaction de Baeyer-Villiger). Ces derniers réagissent avec les aldéhydes et cétones, en donnant un ester et l’acide correspondant (réaction 3). L’hydrolyse de l’ester donne l’acide correspondant à celui des groupements fixés au carbonyle qui présente la moindre aptitude à la migration (méthyl 礪 alkyle primaire 礪 alkyle secondaire, aryle 礪 alkyle tertiaire 礪 hydrogène).DécarboxylationsLes acides 見-cétoniques, l’acide oxalique et les 廓-diacides sont décarbonylés ou décarboxylés par chauffage et conduisent aux acides carboxyliques simples.3. Structure et propriétés physiquesLa structure électronique du groupe carboxylique est caractérisée par la conjugaison interne du système 神, qui a pour conséquence une coplanéité des trois atomes du groupe C2, tous trois hybrides sp2 (fig. 1). Dans l’ion carboxylate, 漣C2-, deux doublets de symétrie p ou 神 sont partagés entre les trois atomes: ils sont décrits par une première orbitale moléculaire liante, de symétrie 神 et par une seconde non liante, antisymétrique par rapport au plan contenant les trois noyaux ainsi que par rapport au plan médiateur de la structure. Une troisième orbitale moléculaire, antiliante, inoccupée dans l’état fondamental (lowest unoccupied molecular orbital , L.U.M.O.), est disponible pour recevoir, en partage, un doublet apporté par un réactif nucléophile. Dans l’acide lui-même (face=F0019 漣C2H), les trois types d’atomes se partagent également deux doublets de symétrie p ou 神; la pondération des orbitales atomiques dans les trois orbitales moléculaires est un peu différente de celle de l’anion, mais la structure électronique est décrite d’une manière comparable.À la structure de l’anion carboxylate, totalement symétrique, correspond une stabilisation, par conjugaison supérieure à celle de la molécule neutre d’acide dont la structure est dissymétrique. Une conséquence est que l’ion carboxylate, stabilisé par conjugaison et fortement solvaté par l’eau, présentera, dans les conditions habituelles, une concentration non négligeable et que la molécule carboxylique aura des propriétés acides. L’augmentation de la densité électronique 神, au niveau de l’oxygène non protoné, confère en outre à ce site un caractère basique. Enfin, l’ensemble des propriétés acides et basiques du groupe fonctionnel est responsable d’une importante association intermoléculaire des acides carboxyliques par formation de liaisons hydrogène. Cette association, le plus souvent bimoléculaire (fig. 2), subsiste en partie à l’état gazeux et en solution diluée non dissociante. Elle modifie les températures de changement d’état solide/liquide et liquide/vapeur: l’acide formique bout à 103 0C et l’acide acétique à 117 0C. Comme dans le cadre des alcanes, on note une alternance des températures de fusion des chaînes paires et impaires.L’acide acétique et l’acide propionique sont des solvants de miscibilité, à la fois aquosolubles et liposolubles . Les acides lourds sont nettement liposolubles, ils possèdent des propriétés amphiphiles . Les molécules de sels alcalins (savons) possèdent une extrémité polaire solvatée par l’eau et une longue chaîne carbonée qui ne l’est pas: en solution aqueuse, elles ont tendance à s’organiser en formant des structures ordonnées sphériques (micelles) au sein desquelles les chaînes non polaires se regroupent en minimisant leur énergie par des interactions de van der Waals tandis que les «têtes» ioniques forment, à l’extérieur de la micelle, une surface solvatée par l’eau. Lorsqu’on introduit un tel savon dans une suspension de gouttelettes d’huile dans de l’eau, les molécules amphiphiles s’organisent à l’interface eau-huile en plaçant leur «tête» hydrophile dans la phase aqueuse et leur «queue» lipophile dans la phase huile. Chaque gouttelette, initialement non miscible à l’eau, est ainsi transformée en une gouttelette dont la surface est devenue hydrophile, donc solvatée par l’eau. Tel est le principe, simplifié, de la «dissolution» des graisses par les savons [cf. DÉTERGENTS].La fonction carboxylique absorbe dans l’ultraviolet (vers 200 nm), à la limite d’utilisation des spectromètres usuels. Leur absorption dans l’infrarouge est caractérisée par une bande large 益OH associée, dans la région de 3 000 cm-1 caractérisant la liaison O 漣H, et une bande assez fine, d’élongation C 漣O, située vers 1 700 cm-1. Le proton acide donne, en résonance magnétique nucléaire (R.M.N.), un signal aux champs très faibles ( 嗀 = de 10 à 12 p.p.m.).4. Propriétés chimiquesLes propriétés chimiques des acides carboxyliques peuvent être rattachées à quatre schémas: la coupure hétérolytique de la liaison O 漣H, la substitution nucléophile du groupe OH, la réactivité de la chaîne carbonée, induite, notamment en 見, par le groupe fonctionnel et les décarboxylations.Réactions de la coupure O size=5漣HLa structure électronique de la fonction carboxylique (fig. 1 a) laisse prévoir une importante polarisation de la liaison O 漣H, tant par le jeu des électronégativités que par la conjugaison interne du système. L’hétérolyse de cette liaison conduit d’autre part à un anion carboxylate, dont la structure symétrique (fig. 1 b) est responsable de la grande stabilité. Les acides carboxyliques présentent en effet, en solution aqueuse, une dissociation qui les classe parmi les acides, faibles il est vrai:
L’hydroxycarbonylation de la soude est l’une des voies industrielles d’accès à l’acide formique. Une variante de ce procédé remplace l’eau par le méthanal et conduit au formiate de méthyle qu’une ammonolyse et une hydrolyse irréversible transforment en formamide et acide formique.L’hydroformylation des oléfines [cf. ALCÈNES], suivie de l’oxydation des aldéhydes formés, est également une voie d’accès aux acides carboxyliques (acides propionique et butyrique). Le principal mode de fabrication de l’acide acétique est aujourd’hui la carbonylation du méthanol (cf. acide ACÉTIQUE).Réactions de préparation avec dégradation de la chaîne carbonéeOxydationDe nombreuses réactions d’oxydation, avec coupure de la chaîne carbonée, conduisent à des acides, stade ultime de l’oxydation ménagée des molécules carbonées.Des procédés industriels, fondés sur l’oxydation, sans catalyseur, d’hydrocarbures, permettent de préparer des mélanges, parfois complexes, de produits d’oxydation où les acides carboxyliques sont majoritaires. Le principal problème est celui de la séparation [cf. ALCANES].Au laboratoire, la coupure oxydante des alcènes, par le permanganate ou le bichromate acides, conduisent à des mélanges d’acides. L’ozonolyse des alcènes est une méthode plus analytique que préparative.Plus intéressante est la coupure oxydante des cétones dans des conditions contrôlées:– coupure des méthylcétones par les halogènes en milieu alcalin. Il se forme une trihalométhylcétone (cf. dérivés CARBONYLÉS) qu’un chauffage en milieu basique transforme en haloforme et sel alcalin de l’acide ayant un carbone de moins que la méthylcétone de départ;– oxydation, avec réarrangement, des dérivés carbonylés sous l’action des peracides (réaction de Baeyer-Villiger). Ces derniers réagissent avec les aldéhydes et cétones, en donnant un ester et l’acide correspondant (réaction 3). L’hydrolyse de l’ester donne l’acide correspondant à celui des groupements fixés au carbonyle qui présente la moindre aptitude à la migration (méthyl 礪 alkyle primaire 礪 alkyle secondaire, aryle 礪 alkyle tertiaire 礪 hydrogène).DécarboxylationsLes acides 見-cétoniques, l’acide oxalique et les 廓-diacides sont décarbonylés ou décarboxylés par chauffage et conduisent aux acides carboxyliques simples.3. Structure et propriétés physiquesLa structure électronique du groupe carboxylique est caractérisée par la conjugaison interne du système 神, qui a pour conséquence une coplanéité des trois atomes du groupe C2, tous trois hybrides sp2 (fig. 1). Dans l’ion carboxylate, 漣C2-, deux doublets de symétrie p ou 神 sont partagés entre les trois atomes: ils sont décrits par une première orbitale moléculaire liante, de symétrie 神 et par une seconde non liante, antisymétrique par rapport au plan contenant les trois noyaux ainsi que par rapport au plan médiateur de la structure. Une troisième orbitale moléculaire, antiliante, inoccupée dans l’état fondamental (lowest unoccupied molecular orbital , L.U.M.O.), est disponible pour recevoir, en partage, un doublet apporté par un réactif nucléophile. Dans l’acide lui-même (face=F0019 漣C2H), les trois types d’atomes se partagent également deux doublets de symétrie p ou 神; la pondération des orbitales atomiques dans les trois orbitales moléculaires est un peu différente de celle de l’anion, mais la structure électronique est décrite d’une manière comparable.À la structure de l’anion carboxylate, totalement symétrique, correspond une stabilisation, par conjugaison supérieure à celle de la molécule neutre d’acide dont la structure est dissymétrique. Une conséquence est que l’ion carboxylate, stabilisé par conjugaison et fortement solvaté par l’eau, présentera, dans les conditions habituelles, une concentration non négligeable et que la molécule carboxylique aura des propriétés acides. L’augmentation de la densité électronique 神, au niveau de l’oxygène non protoné, confère en outre à ce site un caractère basique. Enfin, l’ensemble des propriétés acides et basiques du groupe fonctionnel est responsable d’une importante association intermoléculaire des acides carboxyliques par formation de liaisons hydrogène. Cette association, le plus souvent bimoléculaire (fig. 2), subsiste en partie à l’état gazeux et en solution diluée non dissociante. Elle modifie les températures de changement d’état solide/liquide et liquide/vapeur: l’acide formique bout à 103 0C et l’acide acétique à 117 0C. Comme dans le cadre des alcanes, on note une alternance des températures de fusion des chaînes paires et impaires.L’acide acétique et l’acide propionique sont des solvants de miscibilité, à la fois aquosolubles et liposolubles . Les acides lourds sont nettement liposolubles, ils possèdent des propriétés amphiphiles . Les molécules de sels alcalins (savons) possèdent une extrémité polaire solvatée par l’eau et une longue chaîne carbonée qui ne l’est pas: en solution aqueuse, elles ont tendance à s’organiser en formant des structures ordonnées sphériques (micelles) au sein desquelles les chaînes non polaires se regroupent en minimisant leur énergie par des interactions de van der Waals tandis que les «têtes» ioniques forment, à l’extérieur de la micelle, une surface solvatée par l’eau. Lorsqu’on introduit un tel savon dans une suspension de gouttelettes d’huile dans de l’eau, les molécules amphiphiles s’organisent à l’interface eau-huile en plaçant leur «tête» hydrophile dans la phase aqueuse et leur «queue» lipophile dans la phase huile. Chaque gouttelette, initialement non miscible à l’eau, est ainsi transformée en une gouttelette dont la surface est devenue hydrophile, donc solvatée par l’eau. Tel est le principe, simplifié, de la «dissolution» des graisses par les savons [cf. DÉTERGENTS].La fonction carboxylique absorbe dans l’ultraviolet (vers 200 nm), à la limite d’utilisation des spectromètres usuels. Leur absorption dans l’infrarouge est caractérisée par une bande large 益OH associée, dans la région de 3 000 cm-1 caractérisant la liaison O 漣H, et une bande assez fine, d’élongation C 漣O, située vers 1 700 cm-1. Le proton acide donne, en résonance magnétique nucléaire (R.M.N.), un signal aux champs très faibles ( 嗀 = de 10 à 12 p.p.m.).4. Propriétés chimiquesLes propriétés chimiques des acides carboxyliques peuvent être rattachées à quatre schémas: la coupure hétérolytique de la liaison O 漣H, la substitution nucléophile du groupe OH, la réactivité de la chaîne carbonée, induite, notamment en 見, par le groupe fonctionnel et les décarboxylations.Réactions de la coupure O size=5漣HLa structure électronique de la fonction carboxylique (fig. 1 a) laisse prévoir une importante polarisation de la liaison O 漣H, tant par le jeu des électronégativités que par la conjugaison interne du système. L’hétérolyse de cette liaison conduit d’autre part à un anion carboxylate, dont la structure symétrique (fig. 1 b) est responsable de la grande stabilité. Les acides carboxyliques présentent en effet, en solution aqueuse, une dissociation qui les classe parmi les acides, faibles il est vrai: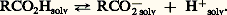 L’acidité est accrue par introduction, dans la chaîne carbonée, de groupes électro-attracteurs: cette propriété est liée à la meilleure dispersion de la charge négative de l’anion sur l’ensemble de la chaîne, ce qui permet une solvatation plus importante de cet anion et entraîne un déplacement de l’équilibre dans le sens de l’ionisation.Les sels alcalins, formés par action des bases, sont solubles, pour les premiers termes. Ils sont thermiquement très stables; l’acétate de sodium anhydre fond sans décomposition à 450 0C. Les sels alcalino-terreux, également solubles pour les premiers termes, sont par contre thermiquement moins stables: vers 300 0C, ils se décomposent en carbonate alcalino-terreux et cétones (cf. dérivés CARBONYLÉS). Les sels d’ammonium, chauffés, se déshydratent, réversiblement, en amides, puis en nitriles.L’hétérolyse de la liaison O 漣H se produit également lors de l’addition des acides carboxyliques sur les alcènes en présence de catalyseur acide avec formation d’un ester:
L’acidité est accrue par introduction, dans la chaîne carbonée, de groupes électro-attracteurs: cette propriété est liée à la meilleure dispersion de la charge négative de l’anion sur l’ensemble de la chaîne, ce qui permet une solvatation plus importante de cet anion et entraîne un déplacement de l’équilibre dans le sens de l’ionisation.Les sels alcalins, formés par action des bases, sont solubles, pour les premiers termes. Ils sont thermiquement très stables; l’acétate de sodium anhydre fond sans décomposition à 450 0C. Les sels alcalino-terreux, également solubles pour les premiers termes, sont par contre thermiquement moins stables: vers 300 0C, ils se décomposent en carbonate alcalino-terreux et cétones (cf. dérivés CARBONYLÉS). Les sels d’ammonium, chauffés, se déshydratent, réversiblement, en amides, puis en nitriles.L’hétérolyse de la liaison O 漣H se produit également lors de l’addition des acides carboxyliques sur les alcènes en présence de catalyseur acide avec formation d’un ester: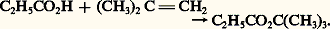 Il en est de même lors de l’estérification des acides par la diazométhane, une méthode douce et spécifique d’obtention des esters méthyliques.Réactions de substitution nucléophile du groupe OHCes réactions de remplacement du groupe OH par un autre nucléophile conduisent aux dérivés de la fonction acide: chlorure d’acide, anhydride, ester, amide. Elles font intervenir, en fait, une addition sur le groupe carbonyle suivie d’une élimination du groupe OH (réaction 4); ce dernier étant un mauvais groupe partant (HO- est en effet basique), il est souvent transformé, au cours de la réaction, en meilleur groupe partant.Formation d’un chlorure d’acide . Opposé à un chlorure d’acide minéral comme PCl3 ou mieux SOCl2, l’acide réagit dans un premier temps en formant un anhydride mixte qui possède un meilleur groupe partant que l’acide de départ. L’ion chlorure attaque cet intermédiaire dans un processus d’addition nucléophile, suivi de l’élimination de molécules neutres (réaction 5).Formation d’un anhydride d’acide . Le nucléophile, dans ce cas, est l’acide lui-même et il faut le modifier pour améliorer son groupe partant; de nombreux réactifs permettent cette modification, notamment des anhydrides d’acides minéraux (P410) ou organique (anhydride acétique en catalyse acide). Comme dans le cas précédent, l’acide substrat réagit, dans un premier temps, en formant avec l’anhydride réactif, activé par le catalyseur acide, un anhydride mixte; une seconde molécule d’acide, jouant le rôle de nucléophile, attaque ce dernier pour former un second anhydride mixte et l’opération se renouvelle une troisième fois avec formation de l’anhydride symétrique. L’ensemble étant réversible, on déplace l’équilibre vers la formation de l’anhydride symétrique en distillant l’acide acétique au fur et à mesure de sa formation (réaction 6) [cf. ANHYDRIDES D’ACIDES CARBOXYLIQUES].Formation d’un ester . L’estérification des acides par les alcools primaires ou secondaires est catalysée par les acides forts. Le catalyseur active l’acide carboxylique en le transformant en son acide conjugué. Le nucléophile alcool attaque ce dernier dans un processus d’addition nucléophile, suivi d’une transprotonation et de l’élimination du bon groupe partant H2O. L’ensemble est réversible et on déplace l’équilibre vers la formation de l’ester soit en mettant en œuvre un large excès de l’alcool, s’il est volatil, soit en éliminant l’eau, notamment par distillation azéotropique [cf. ESTERS].Formation d’un amide . Le nucléophile est l’ammoniac ou encore une amine primaire ou secondaire. On chauffe le sel d’ammonium ou d’aminium qui se déshydrate en amide. En fait, le sel est en équilibre avec l’acide et l’ammoniac, et c’est la réaction entre ces deux partenaires qui conduit, par une addition-élimination, au produit [cf. AMIDES]. Ce même procédé peut être réalisé à température ordinaire grâce à l’intervention d’agents de couplage comme le dicyclohexyl-carbodiimide; cette technique est utilisée en particulier en synthèse peptidique. L’agent de couplage transforme l’acide en anhydride, beaucoup plus réactif, qui est attaqué par la fonction amine et forme la liaison peptidique.Réduction des acides par les hydrures métalliques complexes . Les acides carboxyliques sont facilement réduits en alcools par LiAlH4, dans des conditions douces, à la température ordinaire. Il est possible de stopper la réaction de réduction au stade aldéhyde en traitant l’acide par le métal Li dans NH3 ou CH32, puis hydrolyse de l’imine obtenue.Réactivité de la chaîne carbonée en size=5見En présence de trihalogénures de phosphore, les acides carboxyliques sont halogénés en 見 par les halogènes. La réaction se produit sur l’énol de l’halogénure d’acide, plus stable que celui de l’acide lui-même. Dans un premier temps, l’acide est transformé par PBr3 en bromure d’acide qui, sous sa forme énolique, attaque l’halogène en donnant le bromure d’acide 見-bromé. Ce dernier réalise, avec l’acide, un échange fonctionnel qui conduit à l’acide 見-bromé.Réaction de décarboxylationLa décarboxylation des acides monofonctionnels est difficile. Elle se produit cependant lors de l’électrolyse de leurs sels par l’intermédiaire des radicaux libres carboxyliques formés à l’anode et qui, perdant une molécule de C2, subissent une duplication en alcanes (réaction 7): c’est la réaction de Kolbe.Une autre réaction particulière de décarboxylation concerne les sels d’argent des acides carboxyliques en présence de brome (réaction de Hunsdiecker): on obtient le bromure d’alkyle ayant un carbone de moins que l’acide de départ.5. ApplicationsLes acides carboxyliques présentent une grande importance industrielle. L’acide acétique est l’un des plus importants intermédiaires organiques qui est fabriqué à l’échelle de 5 millions de tonnes par an dans le monde. Pendant longtemps, les acides gras supérieurs comme l’acide stéarique ont servi à la confection des bougies. Leurs sels de sodium ou de potassium constituent les savons courants. Plus récemment, après réduction en alcools correspondants, ces acides gras sont à la base de la préparation de détergents, notamment les sulfates et sulfonates. Les esters des acides moyens ou aromatiques sont employés en parfumerie; de nombreux produits pharmaceutiques dérivent de l’acide benzoïque; enfin, divers sels de l’acide acétique ont des usages particuliers: pigments, mordants, antiseptiques.
Il en est de même lors de l’estérification des acides par la diazométhane, une méthode douce et spécifique d’obtention des esters méthyliques.Réactions de substitution nucléophile du groupe OHCes réactions de remplacement du groupe OH par un autre nucléophile conduisent aux dérivés de la fonction acide: chlorure d’acide, anhydride, ester, amide. Elles font intervenir, en fait, une addition sur le groupe carbonyle suivie d’une élimination du groupe OH (réaction 4); ce dernier étant un mauvais groupe partant (HO- est en effet basique), il est souvent transformé, au cours de la réaction, en meilleur groupe partant.Formation d’un chlorure d’acide . Opposé à un chlorure d’acide minéral comme PCl3 ou mieux SOCl2, l’acide réagit dans un premier temps en formant un anhydride mixte qui possède un meilleur groupe partant que l’acide de départ. L’ion chlorure attaque cet intermédiaire dans un processus d’addition nucléophile, suivi de l’élimination de molécules neutres (réaction 5).Formation d’un anhydride d’acide . Le nucléophile, dans ce cas, est l’acide lui-même et il faut le modifier pour améliorer son groupe partant; de nombreux réactifs permettent cette modification, notamment des anhydrides d’acides minéraux (P410) ou organique (anhydride acétique en catalyse acide). Comme dans le cas précédent, l’acide substrat réagit, dans un premier temps, en formant avec l’anhydride réactif, activé par le catalyseur acide, un anhydride mixte; une seconde molécule d’acide, jouant le rôle de nucléophile, attaque ce dernier pour former un second anhydride mixte et l’opération se renouvelle une troisième fois avec formation de l’anhydride symétrique. L’ensemble étant réversible, on déplace l’équilibre vers la formation de l’anhydride symétrique en distillant l’acide acétique au fur et à mesure de sa formation (réaction 6) [cf. ANHYDRIDES D’ACIDES CARBOXYLIQUES].Formation d’un ester . L’estérification des acides par les alcools primaires ou secondaires est catalysée par les acides forts. Le catalyseur active l’acide carboxylique en le transformant en son acide conjugué. Le nucléophile alcool attaque ce dernier dans un processus d’addition nucléophile, suivi d’une transprotonation et de l’élimination du bon groupe partant H2O. L’ensemble est réversible et on déplace l’équilibre vers la formation de l’ester soit en mettant en œuvre un large excès de l’alcool, s’il est volatil, soit en éliminant l’eau, notamment par distillation azéotropique [cf. ESTERS].Formation d’un amide . Le nucléophile est l’ammoniac ou encore une amine primaire ou secondaire. On chauffe le sel d’ammonium ou d’aminium qui se déshydrate en amide. En fait, le sel est en équilibre avec l’acide et l’ammoniac, et c’est la réaction entre ces deux partenaires qui conduit, par une addition-élimination, au produit [cf. AMIDES]. Ce même procédé peut être réalisé à température ordinaire grâce à l’intervention d’agents de couplage comme le dicyclohexyl-carbodiimide; cette technique est utilisée en particulier en synthèse peptidique. L’agent de couplage transforme l’acide en anhydride, beaucoup plus réactif, qui est attaqué par la fonction amine et forme la liaison peptidique.Réduction des acides par les hydrures métalliques complexes . Les acides carboxyliques sont facilement réduits en alcools par LiAlH4, dans des conditions douces, à la température ordinaire. Il est possible de stopper la réaction de réduction au stade aldéhyde en traitant l’acide par le métal Li dans NH3 ou CH32, puis hydrolyse de l’imine obtenue.Réactivité de la chaîne carbonée en size=5見En présence de trihalogénures de phosphore, les acides carboxyliques sont halogénés en 見 par les halogènes. La réaction se produit sur l’énol de l’halogénure d’acide, plus stable que celui de l’acide lui-même. Dans un premier temps, l’acide est transformé par PBr3 en bromure d’acide qui, sous sa forme énolique, attaque l’halogène en donnant le bromure d’acide 見-bromé. Ce dernier réalise, avec l’acide, un échange fonctionnel qui conduit à l’acide 見-bromé.Réaction de décarboxylationLa décarboxylation des acides monofonctionnels est difficile. Elle se produit cependant lors de l’électrolyse de leurs sels par l’intermédiaire des radicaux libres carboxyliques formés à l’anode et qui, perdant une molécule de C2, subissent une duplication en alcanes (réaction 7): c’est la réaction de Kolbe.Une autre réaction particulière de décarboxylation concerne les sels d’argent des acides carboxyliques en présence de brome (réaction de Hunsdiecker): on obtient le bromure d’alkyle ayant un carbone de moins que l’acide de départ.5. ApplicationsLes acides carboxyliques présentent une grande importance industrielle. L’acide acétique est l’un des plus importants intermédiaires organiques qui est fabriqué à l’échelle de 5 millions de tonnes par an dans le monde. Pendant longtemps, les acides gras supérieurs comme l’acide stéarique ont servi à la confection des bougies. Leurs sels de sodium ou de potassium constituent les savons courants. Plus récemment, après réduction en alcools correspondants, ces acides gras sont à la base de la préparation de détergents, notamment les sulfates et sulfonates. Les esters des acides moyens ou aromatiques sont employés en parfumerie; de nombreux produits pharmaceutiques dérivent de l’acide benzoïque; enfin, divers sels de l’acide acétique ont des usages particuliers: pigments, mordants, antiseptiques.
Encyclopédie Universelle. 2012.